We ordinarily think of the oxidation potential being controlled by the concentrations of the oxidized and reduced forms of a redox couple, as given by the Nernst equation. Under certain circumstances it becomes more useful to think of E as an independent variable that can be used to control the value of Q in the Nernst equation. This usually occurs when two redox systems are present, one being much more concentrated or kinetically active than the other. By far the most important example of this is the way atmospheric oxygen governs the composition of the many redox systems connected with biological activity.
The presence of oxygen in the atmosphere has a profound effect on the redox properties of the aquatic environment— that is, on natural waters exposed directly or indirectly to the atmosphere, and by extension, on organisms that live in an aerobic environment.This is due, of course, to its being an exceptionally strong oxidizing agent and thus a low-lying sink for electrons from most of the elements and all organic compounds.
Those parts of the environment that are protected from atmospheric oxygen are equally important because it is only here that electrons are sufficiently available to produce the "reducing" conditions that are essential for processes varying from photosynthesis to nitrogen fixation.
Electron activity and pE
If we can have pH, why not pE?
As you will recall from your study of acid-base chemistry, the pH of a solution (defined as –log {H+}) is a measure of availablity (technically, the activity) of protons in the solution. As is explained in more detail here, protons tend to "fall" (in free energy) from filled donor levels (acids) to lower acceptor levels (bases.) Through the relation
[H+] ≈ Ka(Ca / Cb)(1-1)
which can be rewritten as
(Ca / Cb) ≈ [H+] / Ka(1-2)
in which the pH is treated as an independent variable that controls the ratio of the conjugate forms of any acid-base pairs in the solution:
log (Ca / Cb) ≈ pH – pKa(1-3)
Electrons, of course, cannot exist as independent particles in aqueous solution, but neither can protons for that matter! It is nevertheless quite valid to refer to the activities of these particles (but not to their "concentrations") when we are considering their availability to donors and acceptors. We will not get into the details of how pE is actually calculated (it is of course related to the ordinary standard electrode potential.)
To get an idea of its significance, consider the following chart that shows the pE° values of some redox systems that are of immense importance in the aquatic environment.
Redox systems and the environment
It's important to bear in mind that the reactions discussed above are mediated by living organisms; without the necessary enzymes to catalyze them, their rates are essentially zero.
For a more detailed chart, see Falling through the respiratory chain.
A very large part of Chemistry is concerned, either directly or indirectly, with determining the concentrations of ions in solution. Any method that can accomplish such measurements using relatively simple physical techniques is bound to be widely exploited. Cell potentials are fairly easy to measure, and although the Nernst equation relates them to ionic activities rather than to concentrations, the difference between them becomes negligible in solutions where the total ionic concentration is less than about 10–3 M.
Electrochemical determination of solubility products
The concentrations of ions in equilibrium with a sparingly soluble salt are sufficiently low that their direct determination can be quite difficult. A far simpler and common procedure is to set up a cell in which one of the electrode reactions involves the insoluble salt, and whose net cell reaction corresonds to the dissolution of the salt. For example, to determine the Ksp for silver chloride, we could use the cell
Ag(s) | Ag+(aq, ? M) || Ag+,Cl– | AgCl(s) | Ag(s)(2-1)
whose net equation corresponds to the dissolution of silver chloride:
cathode: |
AgCl(s) + e– → Ag(s) + Cl–(aq)(2-1a) | E° = +.222 v |
anode: |
Ag(s) → Ag+(aq) + e–(2-1b) | E° = –(+.799) v |
net: |
AgCl(s) → Ag+(aq) + Cl–(aq)(2-1c) | E° = -.577 v |
The standard potential for the net reaction refers to a hypothetical solution in which the activities of the two ions are unity. The cell potential we actually observe corresponds to E in the Nernst equation, which is then solved for Q which gives Ksp directly.
Electrochemistry of potentiometric titrations
In many situations, accurate determination of an ion concentration by direct measurement of a cell potential is impossible due to the presence of other ions and a lack of information about activity coefficients. In such cases it is often possible to determine the ion indirectly by titration with some other ion. For example, the initial concentration of an ion such as Fe2+ can be found by titrating with a strong oxidizing agent such as Ce4+. The titration is carried out in one side of a cell whose other half is a reference electrode:
Pt(s) | Fe2+, Fe3+ || reference electrode(2-2)
Initially the left cell contains only Fe2+. As the titrant is added, the ferrous ion is oxidized to Fe3+ in a reaction that is virtually complete:
Fe2+ + Ce4+ → Fe3+ + Ce3+(2-3)
The cell potential is followed as the Fe2+ is added in small increments. Once the first drop of ceric ion titrant has been added, the potential of the left cell is controlled by the ratio of oxidized and reduced iron according to the Nernst equation
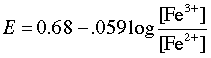 (2-4)
(2-4)
which causes the potential to rise as more iron becomes oxidized.
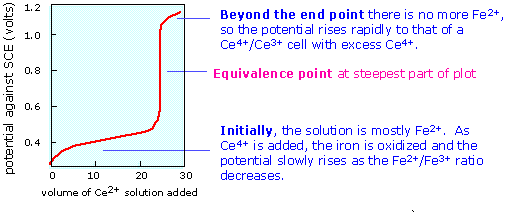
When the equivalence point is reached, the Fe2+ will have been totally consumed (the large equilibrium constant ensures that this will be so), and the potential will then be controlled by the concentration ratio of Ce3+/Ce4+. The idea is that both species of a redox couple must be present in sufficient concentrations to poise an electrode (that is, to control its potential according to the Nernst equation.) If one works out the actual cell potentials for various concentrations of all these species, the resulting titration curve looks much like the familiar acid-base titration curve. The end point is found not by measuring a particular cell voltage, but by finding what volume of titrant gives the steepest part of the curve.
Electrochemical measurement of pH
 Since pH is actually defined in terms of hydrogen ion activity and not its concentration, a hydrogen electrode allows a direct measure of {H+} and thus of –log {H+}, which is the pH. All you need is to measure the voltage of a cell
Since pH is actually defined in terms of hydrogen ion activity and not its concentration, a hydrogen electrode allows a direct measure of {H+} and thus of –log {H+}, which is the pH. All you need is to measure the voltage of a cell
H2(g, 1 atm) | Pt | H+(? M) || reference electrode
In theory this is quite simple, but when it was first employed in the pre-electronics era, it required some rather formidable-looking apparatus (such as the L&N vibrating-reed electrometer setup from the 1920's shown here) and the use of explosive hydrogen gas. Although this arrangement (in which the reference electrode could be a standard hydrogen electrode) has been used for high-precision determinations since that time, it would be impractical for routine pH measurements of the kinds that are widely done, especially outside the research laboratory.
The function of the membrane in the glass electrode is to allow hydrogen ions to pass through and thus change its potential, while preventing other cations from doing the same thing (this selectivity is never perfect; most glass electrodes will respond to moderate concentrations of sodium ions, and to high concentrations of some others.) A glass electrode is thus one form of ion-selective electrode. Since about 1970, various other membranes have been developed which show similar selectivities to certain other ions. These are widely used in industrial, biochemical, and environmental applications.
A comprehensive Beginners Guide to ion-selective electrodes
In 1914 it was discovered that a thin glass membrane enclosing a solution of HCl can produce a potential that varies with the hydrogen ion activity {H+} in about the same way as that of the hydrogen electrode. Glass electrodes are manufactured in huge numbers for both laboratory and field measurements. They contain a built-in Ag-AgCl reference electrode in contact with the HCl solution enclosed by the membrane.
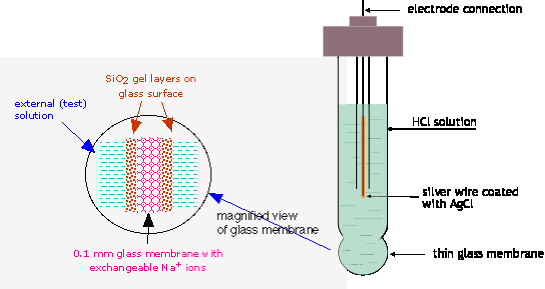
The potential of a glass electrode is given by a form of the Nernst equation very similar to that of an ordinary hydrogen electrode, but of course without the H2:
Emembrane = A + (RT/F) ln ( {H+} + B )
in which A and B are constants that depend on the particular glass membrane.
The reason a glass membrane would behave in this way was not understood until around 1970. It now appears that hydrogen ions in the external solution diffuse through the glass and push out a corresponding number of the Na+ ions which are normally present in most glasses. These sodium ions diffuse to whichever side of the membrane has the lower concentration, where they remain mostly confined to the surface of the glass, which has a porous, gelatinous nature. It is the excess charge produced by these positive ions that gives rise to the pH-dependent potential.
The first commercial pH meter was developed by Arnold Beckman (1900-2004) while he was a Chemistry professor at CalTech. He was unable to interest any of the instrumentation companies in marketing it, so he founded his own company and eventually became a multi-millionaire philanthropist.
 |
 |
 |
| Arnold Beckman | The first commercial Beckman pH meter (1936) | A typical modern pH meter |
You may recall the phenomena of osmosis and osmotic pressure that are observed when two solutions having different solute concentrations are separated by a thin film or membrane whose porosity allows small ions and molecules to diffuse through, but which holds back larger particles.
If one solution contains a pair of oppositely-charged ionic species whose sizes are very different, the smaller ions may pass through the semipermeable membrane while the larger ones are retained. This will produce a charge imbalance between the two solutions, with the original solution having the charge sign of the larger ion.
Eventually the electrical work required to bring about further separation of charges becomes too large to allow any further net diffusion to take place, and the system settles into an equilibrium state in which a constant potential difference (usually around a volt or less) is maintained.
 This potential difference is usually called a membrane potential or Donnan potential after the
English chemist who first described this phenomenon around 1930.
It can be observed simply by introducing a pair of platinum electrodes into the two sides of the cell.
This potential difference is usually called a membrane potential or Donnan potential after the
English chemist who first described this phenomenon around 1930.
It can be observed simply by introducing a pair of platinum electrodes into the two sides of the cell.
The value of this potential difference can be expressed by a relation that is essentially the same as the Nernst equation, although its derivation is different. The membrane potential can be expressed in terms of the ratio of either the K+ or Cl– ion activities:
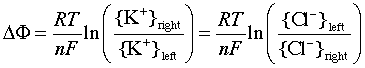
The membrane surrounding most living cells contains sites or "channels" through which K+ ions are selectively transported so that the concentration of K+ inside the cell is 10-30 times that of the intracellular fluid. Taking the activity ratio as about 20, the above equation predicts that the potential difference θinside - θoutside will be
![]()
which is consistent with observed values. Transport of an ion such as K+ from a region of low concentration into the more concentrated intercellular fluid requires a source of free energy, which is supplied by ATP under enzymatic control. The metabolic processes governing this action are often referred to as "ion pumps".
See this site for nice simulations of some of these effects with good explanatory material. (Look under "Diffusion Potentials".)
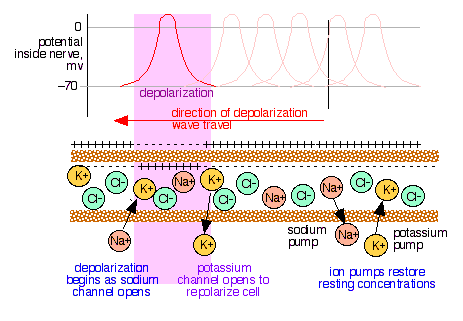
Electrochemistry of nerve conduction
Transmission of signals through the nervous system occurs not by the movement of a charge carrier through the nerve, but by waves of differential ion concentrations that travel along the length of the nerve. These concentration gradients are reduced by protein-based ion channels and ATP-activated (and energy-consuming) ion pumps specific to K+ and Ca2+ ions.